从头计算法(ab initio method)在量子化学计算中指基于量子力学基本原理直接求解薛丁格方程的量子化学计算方法。从头计算法的特点是没有经验参数,并且对体系不作过多的简化。对各种不同的化学体系採用基本相同的方法进行计算。目前的从头计算法包括基于哈特里-福克方程的哈特里–福克方法、在哈特里–福克基础上引入电子相关作用校正而发展起来的后哈特里–福克方法,以及多组态多参考态方法等。与半经验方法相比,从头计算法精度高,但耗时长。
基本介绍
- 中文名:从头计算法
- 外文名:ab initio method
- 作用:直接求解薛丁格方程
- 属性:量子化学计算方法
近似
从头计算法中引入的近似很少,主要包括
- 玻恩–奥本海默近似
- 单电子近似
- 非相对论近似
对于具体的方法而言,可能用到其中的一种或几种近似。
玻恩–奥本海默近似
玻恩–奥本海默近似(Born-Oppenheimer approximation,简称BO近似,又称绝热近似)是一种普遍使用的解包含电子与原子核的体系的量子力学方程的近似方法。
在用量子力学处理分子或其他体系时,需要通过解薛定锷方程或其他类似的偏微分方程获得体系波函式。这个过程往往由于体系自由度过多而非常困难,甚至无法进行。据玻恩-奥本海默近似中,考虑到原子核的质量要比电子大很多,一般要大3-4个数量级,因而在同样的相互作用下,电子的移动速度会较原子核快很多,这一速度的差异的结果是使得电子在每一时刻仿佛运动在静止原子核构成的势场中,而原子核则感受不到电子的具体位置,而只能受到平均作用力。由此,可以实现原子核坐标与电子坐标的近似变数分离,将求解整个体系的波函式的複杂过程分解为求解电子波函式和求解原子核波函式两个相对简单得多的过程。
在玻恩-奥本海默近似下,体系波函式可以被写为电子波函式与原子核波函式的乘积
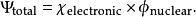
玻恩-奥本海默近似由于在大多数情况下非常精确,又极大地降低了量子力学处理的难度,被广泛套用于分子结构研究、凝聚体物理学、量子化学、化学反应动力学等领域。
玻恩–奥本海默近似是由物理学家奥本海默与其导师玻恩共同提出的。
利用玻恩-奥本海默求解薛定锷方程的步骤
首先将体系的哈密顿算符分解为原子核动能算符与电子哈密顿算符两项
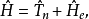
接下来,解以下的电子薛丁格方程以获得电子波函式和绝热势能面:
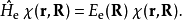
获得势能面之后,可以用其求解原子核的定态薛丁格方程得到原子核波的定态函式和体系能量
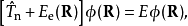
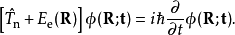
这两种方法获得的结果可以互相转化。原子核定态波函式描述了分子的振动,一般用来获得振动零振动点能,进行红外、微波光谱或电子光谱振动与转动结构的计算;而波函式随时间的演变则描述了化学反应的动力学过程。
注意到第二步原子核薛丁格方程的求解中,需要电子态能量以原子核位置为坐标的函式,因而往往需进行多次,乃至数千万次电子结构的计算,造成计算严重的困难。即使在小分子体系中,往往也无法採用完全量子理学的方法求解波函式。由于原子核的质量比电子大得多,量子效应不如电子明显,因此这一步往往採用比电子结构步骤更高程度的近似方法,以减轻计算负担。例如常採用经典力学或者半经典近似处理原子核运动;在振动幅度较小的情况下,可将势能面近似为二次曲面,而原子核的运动则转化为量子谐振子问题而可解析解出。某些情况下亦可採用平衡态的近似,运用统计力学或过渡态理论等统计方法则,只需要在少量关键位置进行电子结构和振动计算就得到关于分子的分布或反应速率等信息。
适用性
玻恩-奥本海默近似只有在所在电子态与其他电子态能量都足够分离的情况下才有效。当电子态出现交叉或者接近时,玻恩-奥本海默近似即失效。
最低值原理
对于基态,不加其他近似的情况下通过玻恩-奥本海默近似得出的体系总能量一定小于体系真实能量,因而给出了真实能量的下限。与此相对,另一绝热近似方法玻恩-黄近似则给出体系真实能量的上限。
哈特里-福克方程
哈特里-福克方程(英语:Hartree–Fock equation),又称为HF方程,是一个套用变分法计算多电子系统波函式的方程,是量子物理、凝聚态物理学、量子化学中最重要的方程之一。HF方程形式上是单电子本徵方程,求得的本徵态是单电子波函式,即分子轨道。以HF方程为核心的数值计算方法称为“哈特里-福克方法”(Hartree–Fock method)。
基于分子轨道理论的所有量子化学计算方法都是以HF方法为基础的。鑒于分子轨道理论在现代量子化学中的广泛套用,HF方程被视为现代量子化学的基石。
哈特里-福克近似
哈特里-福克近似也称为分子轨道近似或单行列式近似,认为多电子体系波函式可以由体系分子轨道波函式构造的单个斯莱特行列式表示:


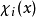
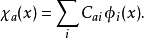
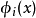
在此近似下,由变分法,体系的多电子薛丁格方程可化为单电子方程
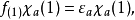
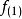
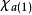
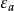
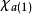
发展简史
1927年,物理学家沃特·海特勒和弗里茨·伦敦完成了氢气分子的量子力学计算之后,开启了量子化学的时代。从此,人们便开始尝试使用量子力学理论来解释化学物质结构和化学现象。
为了解决多电子体系薛丁格方程近似求解的问题,量子化学家道格拉斯·哈特里在1928年提出了哈特里假设,将每个电子看做是在其他所有电子构成的平均势场中运动的粒子,并且首先提出了叠代法的思路。哈特里根据他的假设,将体系电子哈密顿运算元分解为若干个单电子哈密顿运算元的简单代数和,每个单电子哈密顿运算元中只包含一个电子的坐标,因而体系多电子波函式可以表示为单电子波函式的简单乘积,这就是哈特里方程。但是由于哈特里没有考虑电子波函式的反对称要求,他的哈特里方程实际上是非常不成功的。
1930年,哈特里的学生弗拉基米尔·福克和约翰·斯莱特分别提出了考虑泡利原理的自洽场叠代方程和单行列式型多电子体系波函式,这就是哈特里-福克方程。但是由于计算上的困难,HF方程诞生后整整沉寂了二十年,在1950年,量子化学家克莱门斯·罗特汉想到将分子轨道用原子轨道的线性组合来近似展开,而得到了闭壳层结构的罗特汉方程。
1953年,美国的R.帕里瑟、R.帕尔和英国的约翰·波普尔花费两年时间使用手摇计算器分别独立地实现了对氮气分子的RHF自洽场计算,这是人类首次通过求解HF方程获得对化学结构的量子力学解释,也是量子化学计算方法第一次实际完成。在第一次成功之后,伴随着电脑技术的迅猛发展,HF方程与量子化学一道获得长足发展,在HF方程的基础上,人们发展出了高级量子化学计算方法,使得计算精度进一步提高,通过对HF方程电子积分的简化和参数化,人们大大缩减了量子化学的计算量,使得对超过1000个原子的中等大小分子的计算成为可能。
相关条目
- 非绝热耦合
- 薛丁格方程
- 玻恩-黄近似
- 玻恩定则